Во месецот со најмалку дена во годината, во месецот кој и оваа година е редок по своите ретки 28 дена и во месецот кој пациентите со ретки болести го идентификуваат како СВОЈ, по ОСМИ пат ќе се обидеме да се сретнеме со храбрите приказни и поблиску да Ве запознаеме со сите оние кои живеат со предизвиците и проблемите кои се невидливи. Ајде да започнеме нова страница во разбирањето и емпатијата, бидејќи сите заедно ја имаме силата на заедништвото, силата на надежта, силата на информирањето и силата да ги обоиме сенките носејќи светлина и видливост…
КЛИПЕЛ–ФЛЕИЛОВ СИНДРОМ/СИНДРОМ НА ЦЕРВИКАЛНА ФУЗИЈА НА ПРШЛЕНИТЕ/KLIPPEL–FEIL SYNDROME MKБ-10 Q76.1
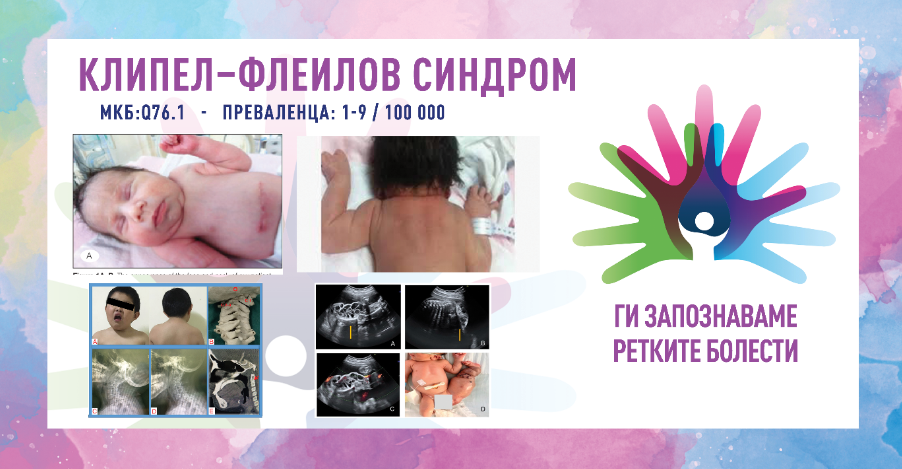 ПРЕВАЛЕНЦА – забележана кај 1/40 000 до 1/42 000 новороденчиња низ светот.
ПРЕВАЛЕНЦА – забележана кај 1/40 000 до 1/42 000 новороденчиња низ светот.
ВОВЕД
Клипел–Флеиловиот синдром, познат и како синдром на цервикална фузија на пршлените, е ретка конгенитална состојба која се карактеризира со абнормална фузија на која било од седумте коски на вратот (вратни пршлени). Може да резултира со ограничена способност за движење и краткост на вратот. Болеста е присутна и се дијагностицира при раѓање, со исклучок на поблаги случаи каде се дијагностицира подоцна при пројавување или влошување на симптомите. Повеќето луѓе имаат само еден или два од тие симптоми кои не се забележливи без медицинска слика. Присутно е искривување на ᾿рбетот – сколиоза, спина бифида – нецелосно формирање на ᾿рбетот за време на бременоста, односно бебето се раѓа со нецелосно споени ᾿рбетни пршлени и други скелетни абнормалности. Нарушувањето, исто така, може да биде поврзано со абнормалности на главата и лицето, скелетот, половите органи, мускулите, мозокот и ᾿рбетниот мозок, рацете, нозете и прстите. Болеста првично била пријавена во 1884 година од страна на Морис Клипел и Андре Феил од Франција. Во 1919 година, во својата докторска теза, Андре Феил предложил друга класификација на синдромот, која опфаќа не само деформација на цервикалниот ᾿рбет, туку и деформација на лумбалниот и торакалниот ᾿рбет.
ЕТИОЛОГИЈА
Кај најголем дел од лицата со Клипел–Флеиловиот синдром, болеста се појавува спорадично, односно од непознати причини. Но, во други случаи постои и наследен фактор, автосомно доминантно или автосомно рецесивно наследување. Мутациите на гените GDF6, GDF3 и MEOX1 се поврзани со синдромот. Мутациите на GDF6, GDF3 и MEOX1 предизвикуваат намален број на функционални протеини кои се кодирани од страна на овие гени, но не е јасно како недостигот на овие протеини доведува до нецелосно одвојување на пршлените кај луѓето со Клипел–Флеиловиот синдром. GDF6 и GDF3 му даваат на телото инструкции за создавање на протеини вклучени во регулирањето на растот и созревањето на коските и ᾿рскавицата. GDF6 конкретно е вклучен во формирањето на вертебралните коски, меѓу другото, и воспоставувањето на граници помеѓу коските во скелетниот развој. GDF3 е вклучен во растот на коските и ᾿рскавицата. Кога синдромот е предизвикан од мутации во гените GDF6 или GDF3, тој се наследува во автосомно доминантен модел, што значи дека една копија од изменетиот ген во секоја клетка е доволна за да предизвика нарушување. Протеинот произведен од генот MEOX1, наречен homeobox-протеин MOX-1, го регулира процесот што започнува со одвојување на пршлените еден од друг за време на раниот развој. Кога овој синдром е предизвикан од мутации во генот MEOX1, тој се наследува во автосомно рецесивен модел, што значи дека и двете копии на генот во секоја клетка имаат мутации. Мутациите можат да се наследат на два начина:
- автосомно доминантно наследување, каде што една копија од изменетиот ген во секоја клетка е доволна за да предизвика нарушување, и е поврзано со фузијата на C2-C3;
- автосомно рецесивно наследување, каде што и двете копии на ген содржат мутации, и е поврзано со фузијата на C5-C6.
Идентификувана е друга автосомно доминантна форма (мапирана на локус 8q22.2), позната како Клипел–Флеилов синдром со малформација на ларинксот.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Клипел–Флеиловиот синдром се јавува во три форми, од кои е познато тека тип 1 се јавува спорадично, а тип 2 се јавува автосомно доминантно, а тип 3 се јавува автосомно рецесивно. Кај тип 1 постои фузија на најголем дел од или на цел цервикален ᾿рбет, кај тип 2, фузијата е на еден или два прешлена од цервикалниот ᾿рбет и кај тип 3 се јавуваат истите промени од тип 1 или тип 2, но придружени со фузија во торакалниот и/или лумбарниот дел од ᾿рбетот. Обично се дијагностицира по раѓањето. Специфичните симптоми варираат од пациент до пациент, но синдромот се карактеризира со тријада симптоми кои вклучуваат ограничена подвижност на вратот и горниот дел од ᾿рбетот, како и пократок врат. Најновите истражувања покажуваат дека оваа тријада ја немаат сите пациенти што значи дека кај некои се манифестира дел од овие симптоми, кај други сите симптоми. Покрај тријадата, други знаци кои се јавуваат се:
- фузија на две или повеќе ᾿рбетни коски во вратот, краток врат и ниско поставена коса;
- тортиколис – главата е наведната на едната страна, а брадата е свртена на другата страна;
- спина бифида – вродено нарушување предизвикано од нецелосно затворање во матката на невралната туба која го обвива ᾿рбетниот мозок;
- главоболки, болки во вратот и појава на испакнување на вратот поради позицијата на скапулата;
- делумно неразделени прсти на раката со изглед кој асоцира на мрежа на пајак (синдактилија), нецелосен развој на скапулата (лопатките на рамото), а со тоа и ипсилатерално ограничени движења на раката;
- синкинезија – движење на огледало, при кое движењето во едната рака неволно го имитира намерното движење на другата рака;
- малформации на бубрезите, ребрата и срцето;
- појава на бубрег во форма на потковица (horse-shoe kidney) кој се развива во ембрионалниот период, односно двата бубрега се спојуваат во еден, па се добива бубрег со форма на потковица;
- респираторни проблеми;
- невролошки дефицити;
- проблеми со сетилото за слух и сетилото за вид.
Дијагностичката евалуација започнува при раѓањето преку длабинска клиничка евалуација, идентификација на карактеристичните симптоми и специјализирани тестови. Дијагностицирањето вклучува РТГ-тестови, компјутеризирана томографија и магнетна резонанца кои се користат за да се идентификуваат отворените простори помеѓу одредени цервикални и други пршлени или други абнормалности во коските и зглобовите. Можат да се направат и други специјализирани тестови за да се идентификуваат другите карактеристични абнормалности поврзани со состојбата, како што се оштетувања на слухот или срцеви мани. Важно е и генетското тестирање, во кое се користи примерок од плунката на детето за да се идентификува постоењето на синдромот, а EOС-технологијата за сликање која создава 3-димензионални модели од две рамни слики, овозможува подобрена дијагноза поради позиционирање. Треба да се внимава на диференцијалната дијагноза која вклучува хируршка историја на спинална фузија, анкилозен спондилитис, јувенилен ревматоиден артритис, фибродисплазија осификанс прогресива и активен или „изгорен“ остеомиелитис.
ПОСЛЕДИЦИ
Прогнозата за повеќето засегнати лица со Клипел–Флеиловиот синдром е добра доколку нарушувањето се третира рано и соодветно. Со тимски лекарски пристап според потребите и тегобите на пациентот, може да се има добра контрола врз здравствената состојба на луѓето кои го имаат овој синдром. Споените пршлени можат да го ограничат опсегот на движење на вратот и грбот, како и да доведат до хронични главоболки и болки во мускулите на вратот и грбот. Лицата со минимална зафатеност на коските често имаат помалку проблеми во споредба со лицата кои имаат само неколку погодени пршлени. Скратениот врат може да предизвика мала разлика во големината и обликот на десната и левата страна на лицето (асиметрија на лицето).
Во помалку од 30 % од случаите, кај лицата со синдромот ќе се појават и срцеви мани. Доколку се присутни овие срцеви мани, тие често доведуваат до скратен животен век, во просек од 35–45 години кај мажите и 40–50 кај жените. Оваа состојба е слична на срцевата слабост забележана кај гигантизмот. Треба да се избегнуваат активности кои можат да го повредат вратот бидејќи можат да придонесат за дополнителни оштетувања. Другите болести поврзани со синдромот можат да бидат фатални ако не се лекуваат навреме или ако доцна се дијагностицираат.
ТРЕТМАН
Третманот се заснова врз управување со најчесто поврзаните симптоми, кои вклучуваат болка во вратот, радикулопатија и/или миелопатија. Основата на третманот за пациенти со болка во вратот се конзервативните, неоперативни мерки. Радикулопатијата се третира со конзервативни интервенции и во одредени случаи може да вклучува спинални инјекции. Појавата на миелопатија поради компресија на ᾿рбетниот мозок типично се припишува на вродената цервикална стеноза, но може да се влоши секундарно при компресија на ᾿рбетниот мозок со комбинација на вертебрални остеофити и/или компресија на меките ткива. Пациентите со екстензивна конгенитална фузија на грлото на матката и/или прекумерно движење на сегмент кој не е споен со движењето, се смета дека се изложени на висок ризик за повреда на ᾿рбетниот мозок. Тие треба да имаат разни секојдневни активности за да ја ограничат таквата потенцијална повреда. Хируршката интервенција може да биде оправдана за специфични случаи каде што прекумерното цервикално или краниовертебрално движење се смета за потенцијално нестабилно и е поврзано со зголемен ризик од повреда на ᾿рбетниот мозок.
OhridNews