Во месецот со најмалку дена во годината, во месецот кој и оваа година е редок по своите ретки 28 дена и во месецот кој пациентите со ретки болести го идентификуваат како СВОЈ, по ОСМИ пат ќе се обидеме да се сретнеме со храбрите приказни и поблиску да Ве запознаеме со сите оние кои живеат со предизвиците и проблемите кои се невидливи. Ајде да започнеме нова страница во разбирањето и емпатијата, бидејќи сите заедно ја имаме силата на заедништвото, силата на надежта, силата на информирањето и силата да ги обоиме сенките носејќи светлина и видливост…
КОКЕЈНОВ СИНДРОМ/ COCKAYNE SYNDROME/ NEILL–DINGWALL SYNDROME MKБ-10 Q87.1
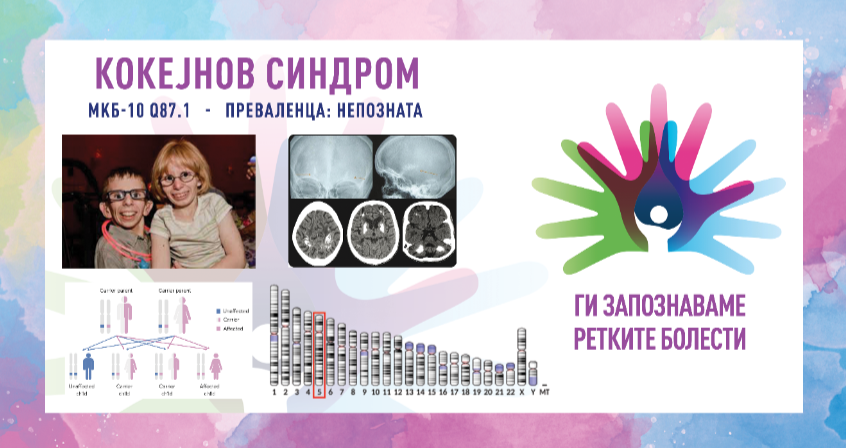 ПРЕВАЛЕНЦА – непозната
ПРЕВАЛЕНЦА – непозната
ВОВЕД
Кокејнов синдром е ретко заболување кое се карактеризира со абнормално мала големина на главата, неуспех за напредок во висина и тежина што доведува до џуџест раст и одложен развој. Заболените со ова нарушување покажуваат уникатни карактеристики на лицето, вклучително и вдлабнати очи, клунест нос и истакнати уши, а исто така доживуваат прогресивна деменција. Знаците и симптомите за оваа состојба обично се видливи уште од детството и тие се влошуваат со текот на времето. Кокејновиот синдром се дели на типови I, II и III врз основа на сериозноста и возраста од почетокот на симптомите.
ЕТИОЛОГИЈА
Синдромот е предизвикан од промени (патогени варијанти) во гените ERCC6 (CSA) и ERCC8 (CSB). Овие гени се вклучени во нормалната поправка на ДНК што се јавува по оштетување од ултравиолетовата светлина и сочинуваат околу 70 % од случаите со генската мутација ERCC6 (CSA). Изложеноста на ултравиолетовата компонента на сончевата светлина, зрачењето и слободните радикали во телото ја оштетува ДНК и бидејќи клетките веќе не се во можност да ја поправат оштетената ДНК, таа се акумулира во клетките. Протеините направени од овие гени се вклучени во поправка на оштетената ДНК преку механизмот за поправка поврзана со транскрипција, особено ДНК во активните гени. Ако генот ERCC6 или ERCC8 е изменет (како кај Кокејнов синдром), оштетувањето на ДНК што се сретнува за време на транскрипцијата, не се поправа, предизвикувајќи РНК-полимеразата да биде позиционирана на таа локација, попречувајќи ја генската експресија. Како што се акумулира несанираното оштетување на ДНК, така е попречена прогресивно-поактивната генска експресија, што доведува до дефект на клетките или клеточна смрт, што веројатно придонесува за знаците на Кокејнов синдром, како што се предвремено стареење и невронска хипомиелинизација. Кокејновиот синдром се наследува автосомно рецесивно и ризикот е ист за машките и женските пациенти.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Карактеристични симптоми на Кокејнов синдром се:
- нарушувања на растот и физичките карактеристики – прогресивното ограничување на растот се јавува на возраст од 1 до 2 години и се карактеризира со неуспех за напредок и микроцефалија; како што стареат децата, тие го губат поткожното масно ткиво, што резултира со прерано стареење на лицето и дисморфни карактеристики, како што се истакнати уши, вдлабнати очи, клунест нос и карактеристично тело со тенки екстремитети;
- невролошки нарушувања и интелектуален развој – невролошките манифестации постепено напредуваат и произлегуваат од демиелинизација; засегнатите деца можат да изразат низа симптоми, вклучително и прогресивно сензоневрално губење на слухот што доведува до длабока глувост, интелектуални попречености, тешкотии при одење, нестабилно одење, лоша рамнотежа, епилепсија во некои случаи, напорен и неразбирлив говор и полиневропатии, притоа најчеста е сензомоторната демиелинизирачка полиневропатија;
- наоди на кожата – тенка и чувствителна кожа, која пациентите ги прави подложни на тешки изгореници од сонце; cупкутаната липоатрофија придонесува за развој на вдлабнати очи и предвремено стареење на прогеричен изглед; вообичаените наоди вклучуваат цијанотични акрални едеми на екстремитетите, дистрофии на ноктите и аномалии на косата;
- абнормалности на забите – мали, неправилни и ненормално распоредени, повеќе склони кон кариес отколку просечната популација;
- мускулно-скелетни – спастичност на нозете, атаксија и контрактури во колковите, колената и глуждовите, издолжени екстремитети, преголеми шаки и стапала;
- окуларни – често се јавуваат пигментирана ретинопатија, оптичка атрофија, миотични зеници, страбизам, катаракта, блефарокератоконјуктивитис и нистагмус;
- ендокрината дисфункција – хипогонадизам кај мажите, кој се јавува во околу 30 % од случаите и неправилна менструација кај жените како резултат на ендокрина дисфункција.
Дијагнозата се поставува врз основа на клиничката слика, клиничките карактеристики и резултатите од тестовите кои во себе вклучуваат:
- генетско тестирање – со помош на примерок до крв се испитува постоењето на мутациите на генот ERCC6 или ERCC8;
- биопсија на кожа;
- МРИ на мозок – може да се открие евентуално постоење на дифузна хипомиелинизација на церебралната бела материја, калцификации во путаменот и вермиска атрофија.
ПОСЛЕДИЦИ
Последиците вклучуваат абнормално висок крвен притисок (хипертензија), бубрежна инсуфициенција, предвремена атеросклероза, гастроезофагеален рефлукс, прогресивна периферна моторна и сензорна невропатија што предизвикува потешкотии при одењето, интелектуална попреченост, доцнење во развојот, неуспех на растот, прогресивна пигментна ретинопатија, катаракта, сензоневрален губиток на слухот, зглобни контрактури, атаксија, тремор, напади, спастичност. Повеќето заболени имаат зголемена чувствителност на сончева светлина и сериозна реакција на антибиотик, наречен метронидазол, како и прерана смрт.
ТРЕТМАН
Децата со сомнение за овој синдром се подложни на мултидисциплинарна евалуација, вклучувајќи клинички преглед, лабораториски и имиџинг-студии, првенствено за да се исклучат другите дијагнози. Сеопфатната евалуација вклучува генетски испитувања, офталмолошки преглед со електроретинографија, невролошки со електроенцефалографија и студии за нервна спроводливост, како и гастроинтестинални, аудиолошки, дерматолошки, стоматолошки и ендокринолошки испитувања.
Третманот на Кокејнов синдром е симптоматски и поддржувачки. Пристапот може да вклучува специјално образование, физикална терапија и други медицински, социјални и/или стручни услуги. Се препорачува генетско советување за членовите на семејството.
Децата со тип I обично одржуваат добри комуникациски вештини и покрај болеста. Се препорачува поттикнување на социјализација и образование соодветно на возраста. Од друга страна, децата со тип II се соочуваат со ограничени можности за интеракција со нивната околина поради сериозноста на сензорните и интелектуалните оштетувања, а смртта обично се случува во првата деценија. Спротивно на тоа, пациентите со тип III покажуваат релативно нормален ран развој, посетувајќи формално образование. Сепак, визуелните и аудитивните оштетувања, заедно со слабоста, се развиваат во адолесценцијата.
OhridNews